Working with aDNA pseuodhaploid samples
Source:vignettes/articles/aDNA_pseudohaploids.Rmd
aDNA_pseudohaploids.RmdThe following article shows how to handle ancient DNA in tidypopgen,
using example data from Lazaridis et al. (2016). First, we will
demonstrate how to handle pseudohaploid data. Then, we will show how to
project ancient DNA data onto modern data in a Principal Components
Analysis, replicating the workflow illustrated in the vignette for the R
package smartsnp. Finally, we will replicate the pairwise
Fst analysis from Lazaridis et al. (2016).
First, we can download the data from the Reich lab website into a temporary directory, and unzip the file.
Download the data
temp_dir <- tempdir()
download_url <- "https://reich.hms.harvard.edu/sites/reich.hms.harvard.edu/files/inline-files/NearEastPublic.tar.gz"
download_path <- file.path(temp_dir, "NearEastPublic.tar.gz")
download.file(download_url, download_path, mode = "wb")
utils::untar(download_path, exdir = temp_dir)Read in the data
Now that our data are downloaded, we can use tidypopgen
to read the data into gen_tibble objects, beginning with
the modern data:
ho_modern <- gen_tibble("./data/NearEastPublic/HumanOriginsPublic2068.geno",
quiet = TRUE,
backingfile = tempfile("test_")
)Followed by the ancient data:
ancient <- gen_tibble("./data/NearEastPublic/AncientLazaridis2016.geno",
quiet = TRUE,
backingfile = tempfile("test_")
)Assigning Pseudohaploid data
Due to the damage associated with ancient DNA samples, genotype datasets of ancient individuals often include pseudohaploid data. Instead of diploid genotypes, low coverage samples are represented by a sampling one allele per site. tidypopgen handles gen_tibbles containing pseudohaploid data by assigning them a specific genotype code (-2) to denote that some individuals are pseudohaplodified. The ploidy of each individual is then stored as 1 (denoting pseudohaploid) or 2 (a diploid individual).
Before we proceed with any analysis, we can therefore use the
function gt_pseudohaploid to assign the ploidy of each
individual in our gen_tibble object (based on whether the
first test_n_loci are all homozygote):
ancient <- gt_pseudohaploid(ancient, test_n_loci = 100000)Once the ploidy has been assigned, pseudohaploid data is handled automatically by functions that are designed to work with pseudohaploid data. Some functions are meaningless to use on pseudohaplodified data, and these functions will return an error.
For example, attempting to run indiv_inbreeding on
pseudohaploid data will tell us that this function only works on diploid
data:
indiv_inbreeding(ancient)
#> Error in stopifnot_diploid(.x): this function only works on diploid dataPCA
For our PCA analysis, we will be projecting ancient European
individuals. Therefore, we need to subset the modern data to only modern
West Eurasian populations. We can select these populations by filtering
the gen_tibble object using the filter()
function from the dplyr package:
west_eurasian_pops <- c(
"Abkhasian", "Adygei", "Albanian", "Armenian", "Assyrian", "Balkar", "Basque",
"BedouinA", "BedouinB", "Belarusian", "Bulgarian", "Canary_Islander",
"Chechen", "Croatian", "Cypriot", "Czech", "Druze", "English", "Estonian",
"Finnish", "French", "Georgian", "German", "Greek", "Hungarian", "Icelandic",
"Iranian", "Irish", "Irish_Ulster", "Italian_North", "Italian_South",
"Jew_Ashkenazi", "Jew_Georgian", "Jew_Iranian", "Jew_Iraqi", "Jew_Libyan",
"Jew_Moroccan", "Jew_Tunisian", "Jew_Turkish", "Jew_Yemenite", "Jordanian",
"Kumyk", "Lebanese_Christian", "Lebanese", "Lebanese_Muslim", "Lezgin",
"Lithuanian", "Maltese", "Mordovian", "North_Ossetian", "Norwegian",
"Orcadian", "Palestinian", "Polish", "Romanian", "Russian", "Sardinian",
"Saudi", "Scottish", "Shetlandic", "Sicilian", "Sorb", "Spanish_North",
"Spanish", "Syrian", "Turkish", "Ukrainian"
)
ho_modern <- ho_modern %>% filter(population %in% west_eurasian_pops)To run a PCA on the modern Eurasian individuals, we need to impute
any missing genotypes. But as we have subset our gen_tibble
object, first we need to update our backingfile:
ho_modern <- gt_update_backingfile(ho_modern,
quiet = TRUE
)And then we can impute, using:
ho_modern <- gt_impute_simple(ho_modern, method = "mean")We should also remove any monomorphic loci from our data, as these
loci are uninformative. Monomorphic markers must always be removed
before a PCA is computed using any of the gt_pca
functions.
To do this, we use the select_loci_if function together
with loci_maf to select only genotypes where MAF is greater
than 0:
ho_modern <- ho_modern %>% select_loci_if(loci_maf(genotypes) > 0)Now we can create our PCA object from the modern data:
modern_pca <- gt_pca_partialSVD(ho_modern, k = 2)We can then use augment to add the PCA coordinates for
each individual to our gen_tibble object:
modern_pca_scores <- augment(x = modern_pca, data = ho_modern, k = 2)And tidy to extract the proportion of explained
variance:
pca_variance <- tidy(modern_pca)Projection
Before projecting the ancient samples, we remove ancient individuals and outgroups that are not of interest:
# Samples to remove:
sample_remove <- c(
"Mota", "Denisovan", "Chimp", "Mbuti.DG", "Altai",
"Vi_merge", "Clovis", "Kennewick", "Chuvash", "Ust_Ishim",
"AG2", "MA1", "MezE", "hg19ref", "Kostenki14"
)
ancient <- ancient %>% filter(!population %in% sample_remove)And now we can project our data using the predict
function:
predicted <- predict(
object = modern_pca,
new_data = ancient,
project_method = "least_squares",
as_matrix = FALSE
)Here, we use the “least_squares” argument to generate a least squares
projection that will be comparable to the approach used in
smartpca (also implemented in the R package
smartsnp).
Finally, we can tidy up our data ready to plot by converting the
predicted object to a data frame and adding the population
names:
The PCA and predicted individuals can then be visualized using
ggplot2 by layering a geom of ancient individuals over
modern individuals, using the same syntax as smartsnp:
ggplot() +
geom_point(
data = modern_pca_scores,
aes(x = .fittedPC1, y = .fittedPC2), alpha = 0.5
) +
geom_point(
data = predicted,
aes(.PC1, .PC2, fill = population, shape = population), size = 3
) +
scale_shape_manual(values = rep(21:25, 100)) +
geom_label_repel(
data = predicted,
aes(.PC1, .PC2, label = population, col = population),
alpha = 0.7, segment.color = "NA"
) +
theme_bw() +
theme(legend.position = "none") +
labs(
x = paste0("PC 1 (", round(pca_variance$percent[1], 2), "%)"),
y = paste0("PC 2 (", round(pca_variance$percent[2], 2), "%)")
)
#> Warning: ggrepel: 247 unlabeled data points (too many overlaps). Consider increasing max.overlaps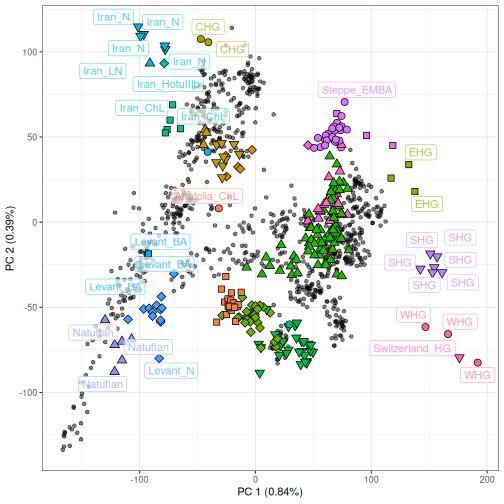
Pairwise Fst
Now let’s replicate the pairwise Fst analysis from Lazaridis et al. (2016).
First we need to merge the ancient and modern data:
ancient_modern <- rbind(ho_modern, ancient)
#> harmonising loci between two datasets
#> flip_strand = FALSE ; remove_ambiguous = FALSE
#> -----------------------------
#> dataset: reference
#> number of SNPs: 548749 reduced to 548749
#> ( 0 are ambiguous, of which 0 were removed)
#> -----------------------------
#> dataset: target
#> number of SNPs: 1233553 reduced to 548749
#> ( 0 were flipped to match the reference set)
#> ( 36301 are ambiguous, of which 36301 were removed)
#>
#> gen_tibble saved to /tmp/RtmpptKMxO/gt_merged_42bc4e3e7eb.gt
#> using bigSNP file: /tmp/RtmpptKMxO/gt_merged_42bc4e3e7eb.rds
#> with backing file: /tmp/RtmpptKMxO/gt_merged_42bc4e3e7eb.bk
#> make sure that you do NOT delete those files!
#> to reload the gen_tibble in another session, use:
#> gt_load('/tmp/RtmpptKMxO/gt_merged_42bc4e3e7eb.gt')Then, we group the ancient and modern data by population:
As we are calculating Fst between populations, we will remove any populations that contain only one individual.
And then we can calculate Fst:
pairwise_fst_tidy <- pairwise_pop_fst(ancient_modern,
method = "Hudson",
type = "tidy"
)We can then assign time ranges to the populations based on their names. This will allow us to group populations by time period when creating our plot:
pairwise_fst_tidy <- pairwise_fst_tidy %>%
mutate(time_range_pop1 = case_when(
str_detect(population_1, "BA") ~ "Bronze Age",
str_detect(
population_1,
"ChL|_EN|_N|Eneolithic"
) ~ "European Neolithic to Chalcolithic",
str_detect(population_1, "HG|Natufian") ~ "Before European Neolithic",
TRUE ~ "Present"
)) %>%
mutate(time_range_pop2 = case_when(
str_detect(population_2, "BA") ~ "Bronze Age",
str_detect(
population_2,
"ChL|_EN|_N|Eneolithic"
) ~ "European Neolithic to Chalcolithic",
str_detect(population_2, "HG|Natufian") ~ "Before European Neolithic",
TRUE ~ "Present"
))After adding the time ranges, we can filter the pairwise Fst data to only include comparisons between populations from the same time range, and set these ranges as factor levels to order the plot:
pairwise_fst_tidy <- pairwise_fst_tidy %>%
filter(time_range_pop1 == time_range_pop2) %>%
mutate(time_range_pop1 = factor(time_range_pop1, levels = c(
"Present",
"Bronze Age",
"European Neolithic to Chalcolithic",
"Before European Neolithic"
)))Finally, we will calculate the median, minimum, and maximum values for each time range, so that these can be added to the plot:
medians <- pairwise_fst_tidy %>%
group_by(time_range_pop1) %>%
summarise(median_value = median(value, na.rm = TRUE)) %>%
arrange(factor(time_range_pop1,
levels = unique(pairwise_fst_tidy$time_range_pop1)
))
min <- pairwise_fst_tidy %>%
group_by(time_range_pop1) %>%
summarise(min_value = min(value, na.rm = TRUE))
max <- pairwise_fst_tidy %>%
group_by(time_range_pop1) %>%
summarise(max_value = max(value, na.rm = TRUE))We can create our plot using ggplot2, adding the median, minimum, and maximum values as lines and text labels:
ggplot(pairwise_fst_tidy, aes(x = value, y = time_range_pop1)) +
geom_point() +
geom_line(
data = medians, aes(x = median_value, y = time_range_pop1, group = 1),
color = "red", linewidth = 1.2
) +
geom_text(data = min, aes(
x = min_value, y = time_range_pop1,
label = round(min_value, 3), vjust = -0.7
)) +
geom_text(data = max, aes(
x = max_value, y = time_range_pop1,
label = round(max_value, 3), vjust = -0.7
)) +
labs(x = expression("F"[ST]), y = element_blank())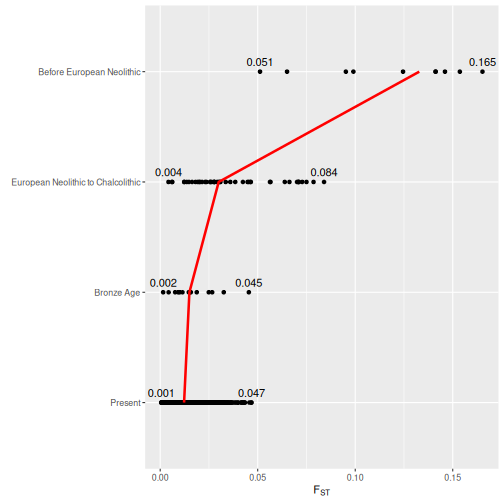
F statistics
To calculate F statistics, tidypopgen integrates with
the R admixtools package (a.k.a. ADMIXTOOLS 2). The
function gt_extract_f2 allows users to calculate blocked f2
statistics from a gen_tibble object, operating in the same
way as the extract_f2 function of
admixtools.
gt_extract_f2 can be used as follows:
f2s <- gt_extract_f2(ancient_modern,
outdir = "./data/NearEastPublic/f2"
)We could now use any of the functions from admixtools
giving the outdir with the f2 values.